A comprehensive guide to pathophysiology, classification, immune mechanisms, diagnosis, and evidence-based management of acute glomerular diseases.
📚 Nephrology · Glomerular Diseases🎓 Medical Education📅 Updated April 2026⏱ 45–60 min read
Table of Contents
- Overview and Clinical Significance
- Glomerular Structure and Function
- Classification of Acute GN
- Pathophysiology
- Clinical Presentation
- Diagnostic Evaluation
- Management
- Prognosis and Outcomes
- Complications
- Clinical Pearls
- Summary
- References
Section 1
Overview and Clinical Significance
Acute glomerulonephritis (AGN) represents a group of kidney diseases that develop rapidly over days to weeks, characterized by inflammation of the glomeruli — the functional filtering units of the kidney. This condition stands as a medical emergency requiring prompt recognition and intervention, as delayed treatment can result in permanent loss of kidney function and progression to end-stage renal disease (ESRD) requiring lifelong dialysis or transplantation.
The hallmark clinical presentation is the acute nephritic syndrome, a distinctive constellation of findings that reflects sudden glomerular injury: visible blood in the urine (hematuria), facial and extremity swelling (edema), elevated blood pressure (hypertension), and declining kidney function (azotemia). The condition affects both children and adults, though the underlying causes and prognosis vary significantly by age and etiology.
Globally, acute GN remains a significant cause of acute kidney injury, particularly in developing nations where post-infectious forms predominate. In developed countries, the spectrum has shifted toward autoimmune and vasculitic forms. Understanding the diverse etiologies, recognizing the clinical presentation, and implementing timely treatment are essential skills for all clinicians managing patients with suspected acute glomerulonephritis.
Clinical Importance: Acute GN can progress to end-stage renal disease within weeks if untreated. Early recognition of the acute nephritic syndrome and urgent diagnostic workup are critical determinants of renal outcomes.
Understanding Glomerular Structure and Function
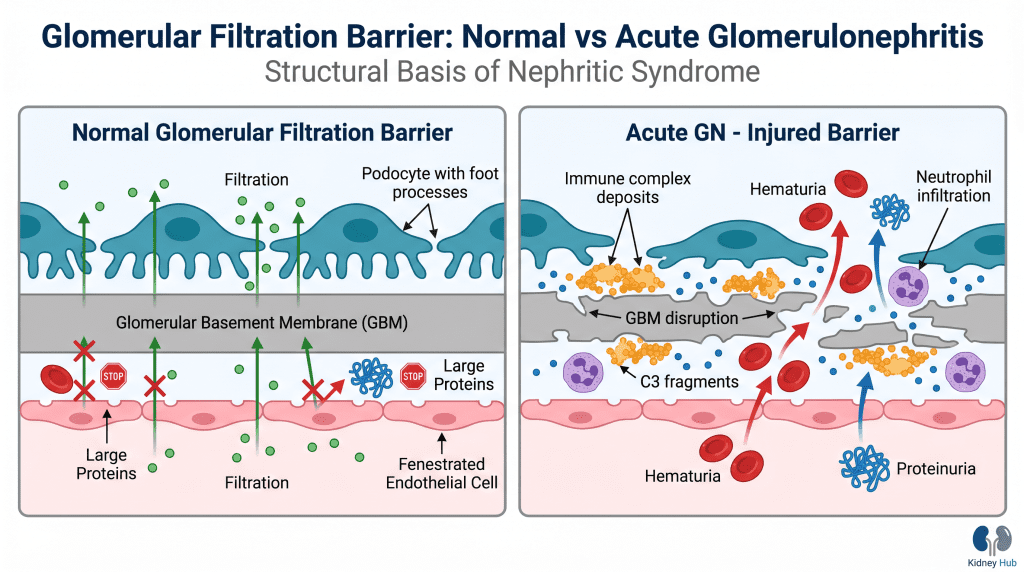
To comprehend how acute glomerulonephritis develops, it is essential to understand normal glomerular anatomy and physiology. The glomerulus consists of several key components working in concert to filter blood while retaining essential proteins and cells.
The glomerular filtration barrier comprises three layers: the fenestrated endothelial cells lining the capillaries, the glomerular basement membrane (GBM) providing structural support, and the podocytes — specialized epithelial cells forming the final filtration barrier with their characteristic foot processes and slit diaphragms. This three-layered structure creates a size- and charge-selective filter that permits passage of water and small solutes while preventing loss of large proteins and blood cells.
When inflammation damages any component of this barrier, selective permeability is compromised. Red blood cells escape into the urine (hematuria), proteins leak across the damaged barrier (proteinuria), and the reduced filtering surface area leads to accumulation of waste products in the blood (azotemia). Inflammatory mediators simultaneously trigger changes in blood vessel tone and fluid handling, resulting in sodium and water retention that manifests as hypertension and edema.
Classification of Acute Glomerulonephritis
Acute glomerulonephritis is organized into distinct categories based on the underlying immunological mechanism driving glomerular injury. This classification is crucial because it guides diagnostic testing, predicts prognosis, and determines treatment strategies. The three principal immunopathological mechanisms are immune complex deposition, pauci-immune neutrophil-mediated injury, and direct anti-GBM antibody binding.
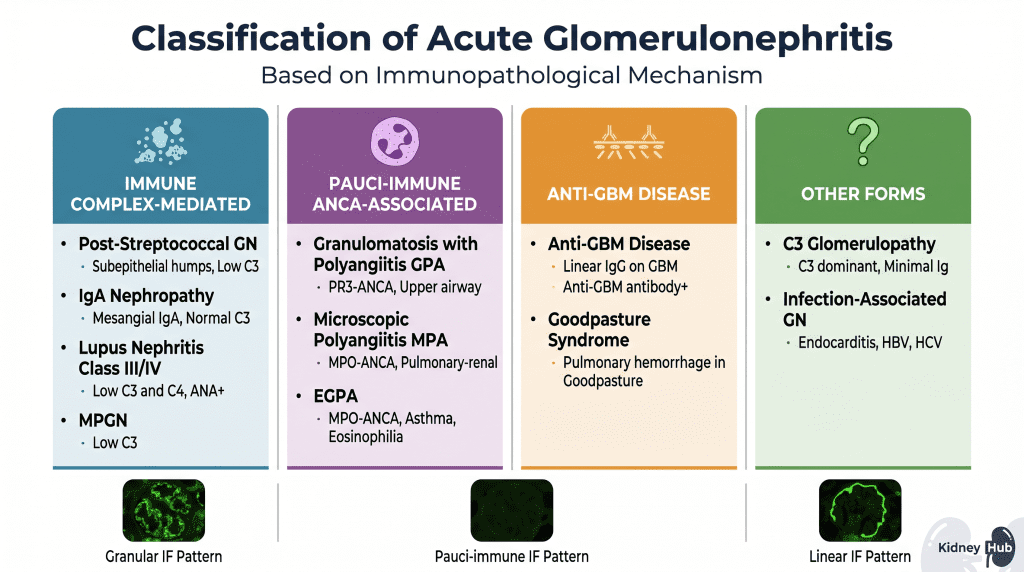
Immune Complex-Mediated Acute GN
In this category, the primary pathogenic mechanism involves deposition of immune complexes — aggregates of antibodies bound to antigens — within the glomerulus. These complexes activate the complement system, triggering an inflammatory cascade that damages glomerular structures. Immunofluorescence reveals a granular pattern of IgG, IgM, and/or IgA with complement deposition.
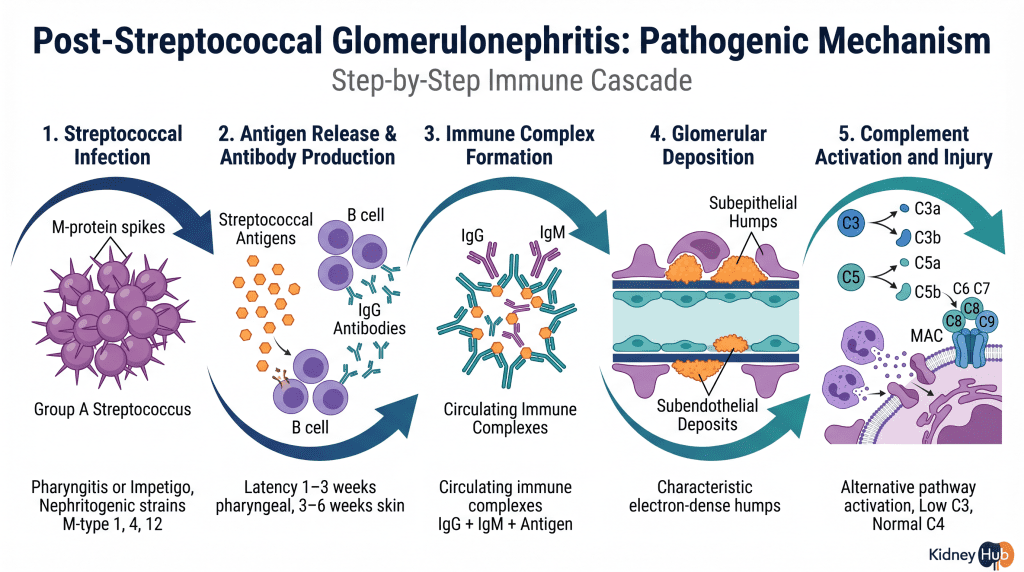
Post-Streptococcal Glomerulonephritis (PSGN)
PSGN represents the most common form of acute GN worldwide, particularly in pediatric populations. This disease follows infection with specific nephritogenic strains of Group A Streptococcus, typically from pharyngitis or impetigo. The characteristic feature is a latency period of 1–3 weeks (pharyngeal) or 3–6 weeks (skin) between the initial infection and onset of kidney disease. During this interval, the immune system generates antibodies against streptococcal antigens — particularly nephritis-associated plasmin receptor (NAPlr) and streptococcal pyrogenic exotoxin B (SPEB) — and these antibodies form complexes that deposit in the glomerulus. The alternative complement pathway becomes activated, recruiting inflammatory cells and triggering glomerular damage.
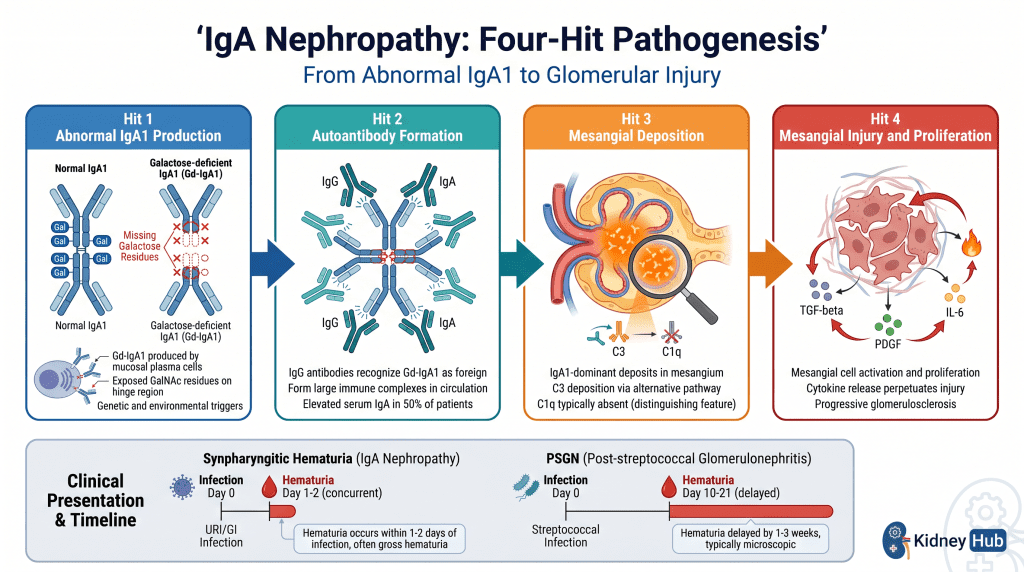
IgA Nephropathy (IgAN)
IgA nephropathy is the most frequently diagnosed primary glomerulonephritis worldwide. The disease is characterized by predominant deposition of immunoglobulin A (IgA) in the glomerular mesangium. Patients produce abnormal, galactose-deficient IgA1 (Gd-IgA1) molecules that are prone to aggregation and complex formation. A distinctive clinical feature is synpharyngitic hematuria — gross hematuria occurring concurrently with (not weeks after) upper respiratory tract infections — because the IgA response is triggered directly by mucosal antigens encountered during the infection.
{kind=link}
Lupus Nephritis (Class III/IV)
Lupus nephritis occurs in the context of systemic lupus erythematosus (SLE), an autoimmune disease affecting multiple organ systems. In acute lupus nephritis, circulating immune complexes containing anti-dsDNA antibodies deposit throughout the glomerulus — in the mesangium, subendothelial, and subepithelial spaces — triggering complement activation via the classical pathway. The acute proliferative forms (Class III: focal, and Class IV: diffuse) present with features of the acute nephritic syndrome and carry the greatest risk of progression to ESRD.
Membranoproliferative GN (MPGN)
MPGN is characterized by mesangial proliferation and thickening of the glomerular basement membrane, resulting from complement dysregulation or immune complex deposition. It can present acutely with nephritic features and is associated with low C3 levels due to persistent complement activation.
Pauci-Immune GN — ANCA-Associated Vasculitis (AAV)
This category encompasses diseases driven by anti-neutrophil cytoplasmic antibodies (ANCA) that bind to antigens on the surface of activated neutrophils, triggering their activation and degranulation. The release of toxic enzymes and reactive oxygen species causes direct glomerular damage, often resulting in crescent formation and rapidly progressive kidney failure. Immunofluorescence reveals a pauci-immune pattern — minimal or absent immunoglobulin and complement deposition — distinguishing this category from immune complex-mediated disease.
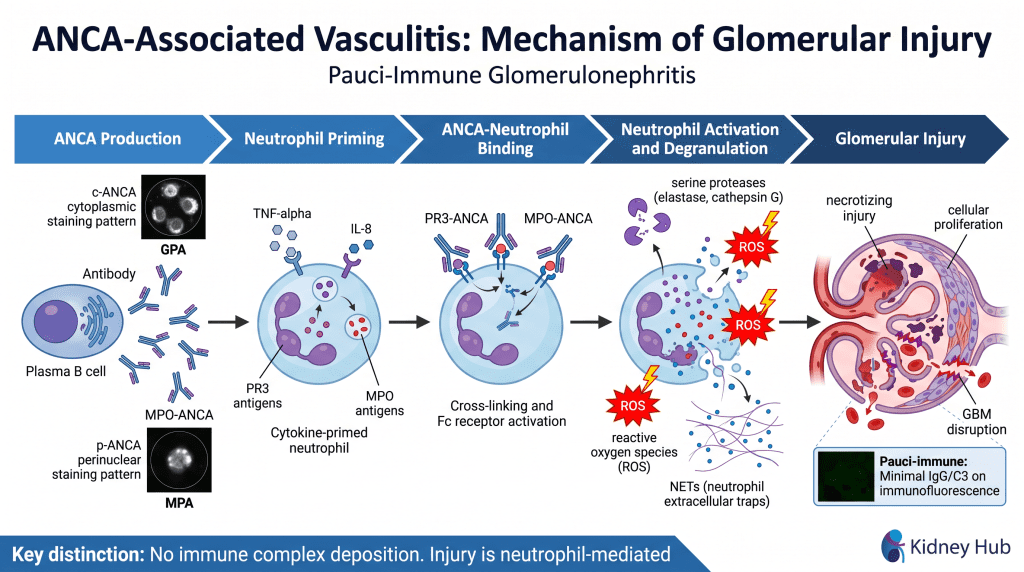

Granulomatosis with Polyangiitis (GPA)
Associated with PR3-ANCA (c-ANCA pattern). Characteristic involvement of upper respiratory tract (sinusitis, nasal granulomas), lungs (pulmonary nodules, hemorrhage), and kidneys. Formerly known as Wegener’s granulomatosis.
Microscopic Polyangiitis (MPA)
Associated with MPO-ANCA (p-ANCA pattern). Typically presents with pulmonary-renal syndrome — simultaneous pulmonary hemorrhage and glomerulonephritis — without upper respiratory granulomas.
Eosinophilic GPA (EGPA)
Combines asthma, peripheral eosinophilia, and systemic vasculitis. MPO-ANCA is frequently present. Cardiac involvement is a major cause of morbidity. Formerly known as Churg-Strauss syndrome.
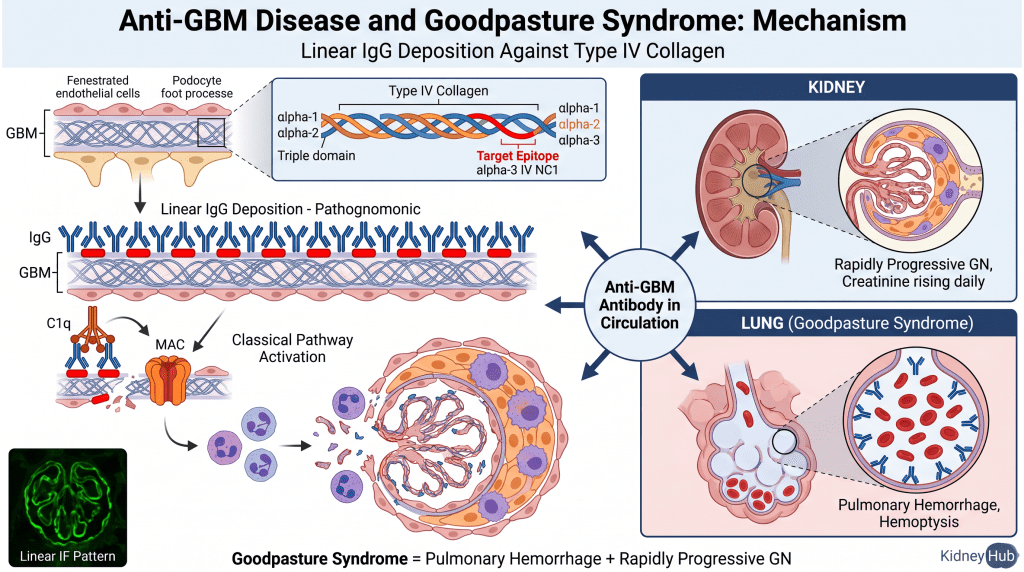
Anti-Glomerular Basement Membrane (Anti-GBM) Disease
This rare but severe form of acute GN results from autoantibodies directed against the non-collagenous domain (NC1) of the alpha-3 chain of type IV collagen — a major structural component of the GBM. These antibodies bind directly to the basement membrane in a linear pattern along its entire length, which is pathognomonic for this disease. When anti-GBM antibodies also bind to the alveolar basement membrane in the lungs, the condition is termed Goodpasture’s syndrome, presenting with pulmonary hemorrhage simultaneously with rapidly progressive glomerulonephritis — a true medical emergency.

Other Forms of Acute GN
C3 Glomerulopathy results from dysregulation of the alternative complement pathway, often due to genetic mutations affecting complement regulatory proteins (e.g., Factor H, Factor I, C3 convertase mutations) or acquired inhibitors (e.g., C3 nephritic factor). This leads to uncontrolled complement activation and C3-dominant deposition in the glomerulus with minimal immunoglobulin deposition — distinguishing it from immune complex-mediated disease.
Infection-Associated GN can follow various infections beyond streptococci, including bacterial endocarditis (often staphylococcal), hepatitis B and C, HIV infection, syphilis, and malaria. In each case, microbial antigens or host antigens modified by infection trigger immune complex formation and glomerular deposition. Unlike PSGN, the infection is often still active at the time of GN presentation.
| Disease | Mechanism | IF Pattern | C3 | C4 | Key Antibody |
|---|---|---|---|---|---|
| PSGN | Immune complex | Granular (C3-dominant) | ↓↓ | Normal | ASO, Anti-DNase B |
| IgA Nephropathy | Immune complex | Mesangial IgA-dominant | Normal | Normal | Elevated serum IgA (50%) |
| Lupus Nephritis | Immune complex | Granular “full house” | ↓↓ | ↓↓ | ANA, Anti-dsDNA |
| ANCA Vasculitis | Pauci-immune | Pauci-immune (minimal) | Normal | Normal | PR3-ANCA or MPO-ANCA |
| Anti-GBM Disease | Anti-GBM antibody | Linear IgG along GBM | Normal | Normal | Anti-GBM antibody |
| C3 Glomerulopathy | Complement dysregulation | C3-dominant, minimal Ig | ↓↓ | Normal | C3 nephritic factor |
Section 4
Pathophysiology of Acute Glomerulonephritis
Mechanisms of Glomerular Injury
Antigen Presentation and Immune Activation
The initial event in most forms of acute GN involves presentation of foreign or self-antigens to the immune system. In PSGN, streptococcal antigens are presented to B cells and T cells, triggering antibody production and T cell-mediated immunity. In IgA nephropathy, abnormally glycosylated IgA1 molecules are recognized as antigenic. In ANCA-associated vasculitis, the immune system produces antibodies against neutrophil cytoplasmic antigens (PR3 and MPO). In anti-GBM disease, the immune system generates antibodies against basement membrane type IV collagen.
Immune Complex Formation and Deposition
In immune complex-mediated diseases, circulating antibodies bind to antigens, forming immune complexes that circulate in the bloodstream and eventually deposit in the glomerulus. Deposition occurs through passive trapping as complexes pass through glomerular capillaries, or through active mechanisms involving interactions with glomerular structures. In some diseases, antigens deposit first in the glomerulus, and antibodies subsequently bind to these “planted” antigens, forming immune complexes in situ. The size and composition of immune complexes influence their deposition pattern: large complexes tend to deposit in the subepithelial space (producing the characteristic “humps” of PSGN), while smaller complexes deposit subendothelially or in the mesangium.
Complement Activation Cascades
Immune complex deposition triggers activation of the complement system, a cascade of plasma proteins that amplify inflammation. Three pathways converge on a common effector mechanism:
The classical pathway is activated when IgG or IgM antibodies bind to antigens. C1q recognizes the antibody-antigen complex and initiates a proteolytic cascade generating C3 and C5 convertases. The alternative pathway is activated directly by certain microbial surfaces and IgA-containing immune complexes, bypassing early components and directly amplifying C3 activation. The lectin pathway is activated when mannose-binding lectin (MBL) recognizes carbohydrate patterns on pathogen surfaces, converging with the classical pathway at C3 activation.
Regardless of the initiating pathway, complement activation generates critical mediators: C3a and C5a act as potent chemoattractants drawing neutrophils and macrophages into the glomerulus; the C5b-9 membrane attack complex forms pores in the GBM; and C3b fragments mark glomerular structures for destruction by cells bearing complement receptors.
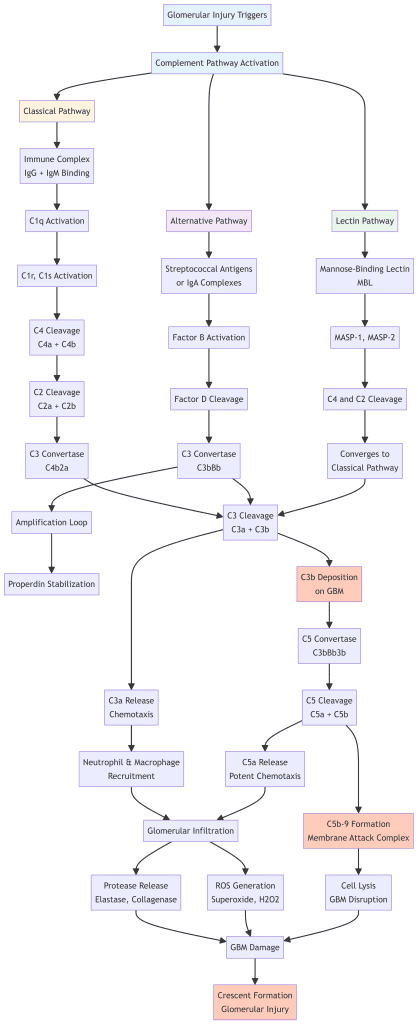
Cellular Infiltration and Mediator Release
Chemoattractant fragments generated by complement activation recruit inflammatory cells into the glomerulus. Neutrophils and macrophages accumulate within glomerular capillaries and surrounding tissue. These cells release numerous harmful mediators including proteases (elastase, cathepsin G), reactive oxygen species (ROS), and cytokines (IL-1β, TNF-α, IL-6). In ANCA-associated vasculitis, this mechanism is the primary driver of injury: ANCA antibodies bind to surface antigens on primed neutrophils, triggering massive degranulation and neutrophil extracellular trap (NET) formation, causing severe glomerular damage in the absence of immune complex deposition.
Crescent Formation and Glomerular Scarring
In severe acute GN, particularly in rapidly progressive forms, intense inflammation causes necrosis of glomerular structures. The parietal epithelial cells lining Bowman’s capsule respond by proliferating, forming crescents — crescent-shaped accumulations of cells that compress the glomerular tuft and obliterate the capillary lumen. In the most severe cases, these crescents undergo fibrosis, leading to permanent scarring and irreversible loss of glomerular function. Crescents in more than 50% of glomeruli define rapidly progressive glomerulonephritis (RPGN) and predict poor renal outcomes without urgent treatment.
Systemic Consequences of Glomerular Injury
Reduction in Glomerular Filtration Rate
The inflammatory process reduces the surface area available for filtration through several mechanisms: proliferation of glomerular cells increases cellularity and reduces capillary lumen; crescent formation physically compresses the glomerular tuft; and swelling of endothelial cells narrows capillary lumens. Serum creatinine and blood urea nitrogen rise as waste products accumulate. In severe cases, GFR falls so dramatically that patients develop oliguria (<400 mL/day) or anuria.
Sodium and Water Retention
Despite reduced GFR, the kidneys retain sodium and water excessively in acute GN. Reduced GFR means less sodium is filtered and excreted. Inflammatory mediators and reduced renal perfusion activate the renin-angiotensin-aldosterone system (RAAS), increasing sodium reabsorption. Sympathetic nervous system activation further enhances sodium reabsorption. The result is positive sodium and water balance, leading to extracellular fluid volume expansion that manifests as hypertension and edema.
Electrolyte Abnormalities
The reduced GFR impairs excretion of potassium, leading to potentially life-threatening hyperkalemia. Phosphate retention leads to hyperphosphatemia, triggering secondary hyperparathyroidism and hypocalcemia. Metabolic acidosis develops as the kidneys fail to excrete hydrogen ions and regenerate bicarbonate — all consequences of the acute reduction in functional nephron mass.Section 5
Clinical Presentation
The Acute Nephritic Syndrome
The classic presentation of acute GN is the acute nephritic syndrome, a distinctive clinical pattern that should immediately alert clinicians to the possibility of glomerular disease. Recognition of this constellation is the essential first step in the diagnostic pathway.

Hematuria
Hematuria is the most common presenting symptom. Patients may report gross hematuria — visible blood in the urine appearing tea-colored, cola-colored, or frankly red — or hematuria may be detected only on urinalysis. The presence of red blood cell casts in the urine is highly specific for glomerular hematuria and is pathognomonic for glomerulonephritis. These casts form when red blood cells become trapped in a protein matrix (Tamm-Horsfall protein) within the renal tubules. Dysmorphic red blood cells — with irregular shapes caused by passage through damaged glomeruli — are another marker of glomerular bleeding.
Proteinuria
Protein loss typically ranges from 0.5 to 3 grams per day in acute GN — significant but usually below the nephrotic range (>3.5 g/day). The proteinuria results from increased glomerular permeability due to inflammatory damage. Patients may notice foamy urine from the high protein content creating surface tension. Nephrotic-range proteinuria can occur in severe forms, particularly lupus nephritis Class IV.
Hypertension
Elevated blood pressure is present in 50–80% of patients with acute GN, developing over days to weeks as sodium and water retention progresses. In some patients, hypertension is mild and asymptomatic. In others, particularly those with severe glomerular injury, blood pressure elevation can be dramatic and lead to hypertensive emergencies with encephalopathy, seizures, or acute heart failure.
Edema
Swelling develops as fluid accumulates in the interstitial space. Periorbital edema — puffiness around the eyes — is particularly characteristic and often the first sign that prompts patients to seek medical attention, especially in children. Peripheral edema affecting the hands, feet, and legs also develops. In severe cases, ascites and pleural effusions may occur.
Azotemia
Elevated serum creatinine and blood urea nitrogen reflect reduced GFR. The degree of azotemia correlates with the severity of glomerular injury. In mild cases, creatinine may be only mildly elevated. In severe cases with extensive crescent formation, creatinine can rise rapidly over days to weeks — the hallmark of rapidly progressive glomerulonephritis (RPGN), which demands urgent evaluation and treatment.
Etiology-Specific Clinical Features
Post-Streptococcal GN
Typically children 5–12 years. History of recent pharyngitis or impetigo. Latency period 1–3 weeks (pharyngeal) or 3–6 weeks (skin). Full nephritic syndrome. Excellent prognosis in children (>95% complete recovery).
IgA Nephropathy
Synpharyngitic hematuria (concurrent with URI, not delayed). Recurrent episodes of gross hematuria. Can present at any age. Slow progression over years to decades. No specific serological marker — biopsy required.
Lupus Nephritis
Young women of childbearing age. Systemic features: malar rash, arthritis, serositis, photosensitivity. Low C3 and C4. ANA and anti-dsDNA positive. Class III/IV presents with acute nephritic syndrome.
ANCA Vasculitis
Systemic symptoms: fever, weight loss, arthralgias. GPA: sinusitis, nasal granulomas, saddle-nose deformity. MPA: pulmonary hemorrhage. Rapidly progressive course — creatinine rising daily. ANCA positive in ~90%.
Anti-GBM Disease
Dramatic presentation. Rapidly progressive GN — creatinine doubling over days. Goodpasture syndrome: hemoptysis + rapidly progressive GN. Bimodal age distribution (20s and 60s). Medical emergency.Section 6
Diagnostic Evaluation
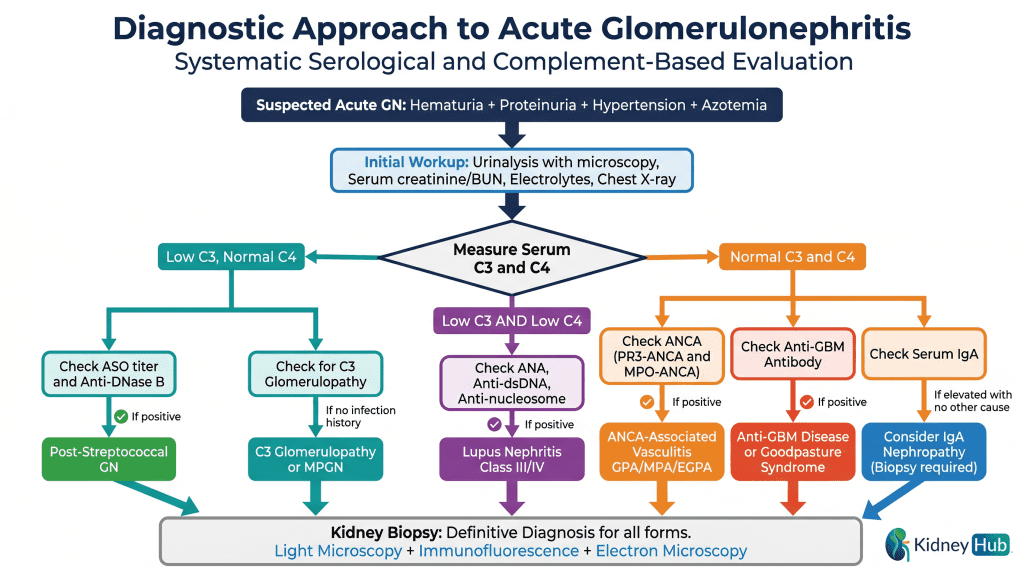
The diagnostic evaluation of suspected acute GN follows a systematic approach, beginning with clinical assessment and urinalysis, proceeding to targeted serological testing guided by complement levels, and culminating in kidney biopsy for definitive diagnosis and histological characterization.
Urinalysis and Urine Microscopy
Urinalysis is the most important initial test for suspected glomerulonephritis. The presence of hematuria with red blood cell casts is highly specific for glomerular disease. Urine should be examined microscopically to identify casts, dysmorphic red blood cells, and white blood cells. Quantification of proteinuria through spot urine protein-to-creatinine ratio or 24-hour urine collection provides information about the degree of protein loss.
Complement Studies
Measurement of serum C3 and C4 levels is essential for differential diagnosis and serves as the initial branch point in the diagnostic algorithm:
- Low C3, Normal C4: Alternative pathway activation — suggests PSGN, C3 glomerulopathy, or MPGN
- Low C3 AND Low C4: Classical pathway activation — suggests lupus nephritis
- Normal C3 and C4: Minimal complement involvement — suggests IgAN, ANCA vasculitis, or anti-GBM disease
Serological Testing
| Disease | Key Serological Tests | Sensitivity | Specificity |
|---|---|---|---|
| PSGN | ASO titer, Anti-DNase B, Throat culture | ASO: 70–80% | High for recent strep infection |
| Lupus Nephritis | ANA, Anti-dsDNA, Anti-nucleosome, C3, C4 | ANA: >95% | Anti-dsDNA: high specificity |
| ANCA Vasculitis | PR3-ANCA (c-ANCA), MPO-ANCA (p-ANCA) | ~90% systemic, 50–80% renal-limited | High when combined with clinical features |
| Anti-GBM Disease | Anti-GBM antibody (ELISA or RIA) | >90% active disease | Very high |
| IgA Nephropathy | Serum IgA (elevated in 50%) | Low — biopsy required | Non-specific |
Kidney Biopsy
Kidney biopsy provides definitive diagnosis and assessment of disease severity. Three modalities are used in combination: light microscopy reveals the pattern of glomerular injury (proliferative, necrotizing, crescent formation); immunofluorescence microscopy identifies the pattern of immunoglobulin and complement deposition, which is pathognomonic for specific diseases; and electron microscopy reveals the ultrastructure of immune deposits and glomerular damage at the nanometer level.
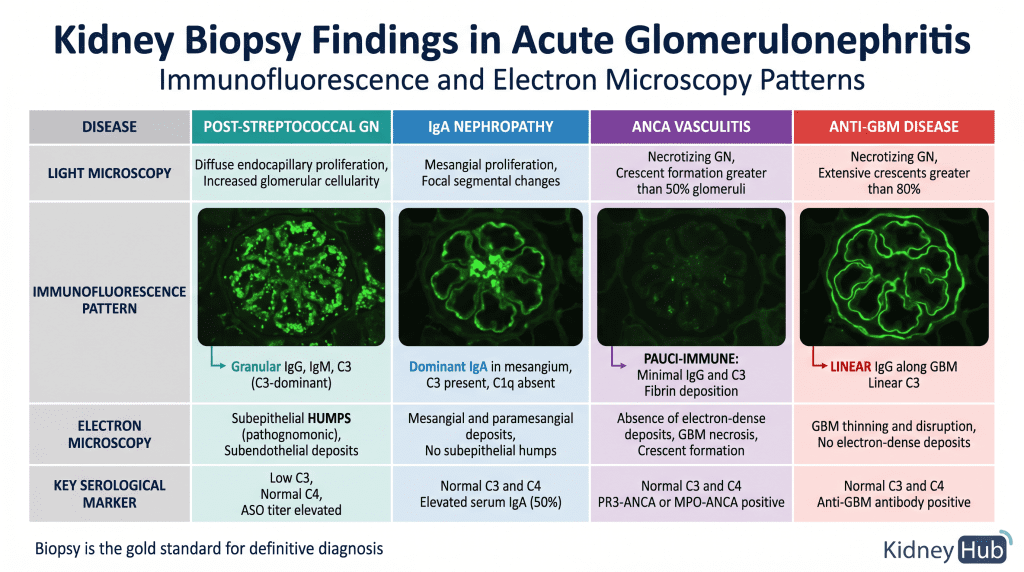
Section 7
Management of Acute Glomerulonephritis
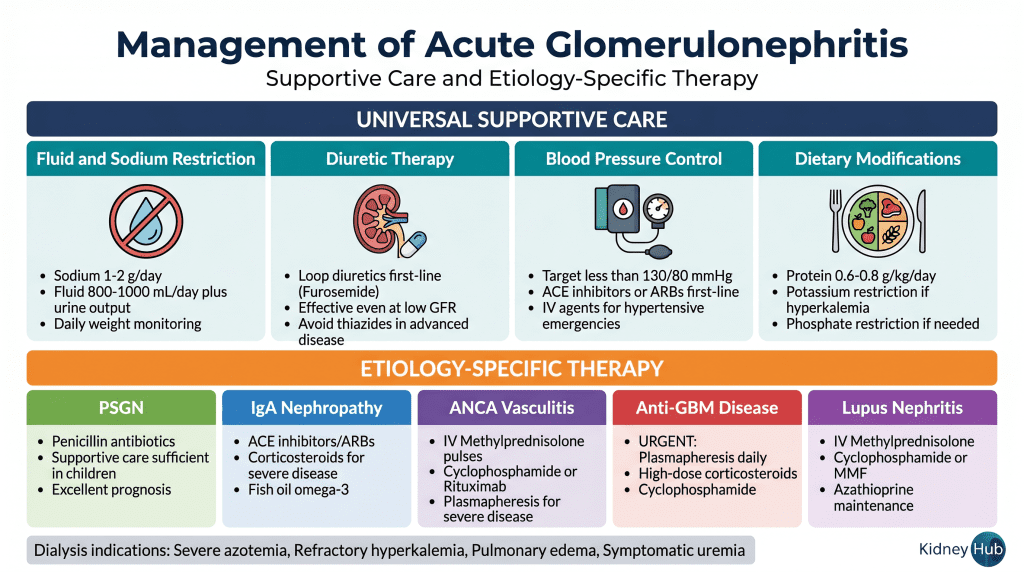
Universal Supportive Care
Fluid and Sodium Restriction
Sodium intake should be restricted to 1–2 grams per day to minimize sodium retention and reduce blood pressure elevation. Fluid intake should be restricted to approximately 800–1000 mL per day plus ongoing urine output losses. Daily weights provide the most accurate assessment of fluid status — a weight gain exceeding 2–3 pounds per day suggests excessive fluid retention requiring more aggressive diuretic therapy.
Diuretic Therapy
Loop diuretics such as furosemide are the first-line agents for managing fluid overload and edema. These agents inhibit sodium reabsorption in the thick ascending limb of the loop of Henle and remain effective even when GFR is reduced. Thiazide diuretics are less effective in advanced renal dysfunction and should generally be avoided as monotherapy.
Blood Pressure Management
Target blood pressure should be less than 130/80 mmHg. ACE inhibitors and angiotensin receptor blockers (ARBs) are preferred first-line agents because they provide renal protection through hemodynamic effects (dilation of the efferent arteriole, reducing intraglomerular pressure) and anti-inflammatory effects. NSAIDs should be avoided as they can worsen renal function. For hypertensive emergencies, intravenous agents such as labetalol, hydralazine, or nicardipine should be used with careful blood pressure monitoring.
Dietary Modifications
Protein intake should be restricted to 0.6–0.8 g/kg/day when azotemia is present, reducing the nitrogen load the kidneys must excrete. Potassium restriction is necessary if serum potassium is elevated. Phosphate restriction may be required if hyperphosphatemia develops. Adequate caloric intake must be maintained to prevent catabolism.
Etiology-Specific Therapy
Post-Streptococcal GN
Antibiotics (penicillin or alternatives) should be administered to eliminate any residual streptococcal infection. However, antibiotics do not alter the course of the glomerulonephritis itself, which is driven by immune complexes already formed. Immunosuppressive therapy is rarely needed in children with uncomplicated PSGN, as most recover completely with supportive care alone. In adults with severe disease or rapidly progressive course, corticosteroids may be considered.
IgA Nephropathy
Supportive care with blood pressure control using ACE inhibitors or ARBs forms the foundation of treatment, slowing the progression of proteinuria and GFR decline. For patients with severe disease, rapidly progressive course, or persistent proteinuria despite ACE inhibitor/ARB therapy, immunosuppressive therapy may be beneficial. Corticosteroids (oral or IV pulses) and mycophenolate mofetil are commonly used. Fish oil (omega-3 fatty acids) may provide additional benefit in slowing disease progression.
ANCA-Associated Vasculitis
High-dose corticosteroids are the cornerstone of induction therapy. IV methylprednisolone pulses (500 mg–1 g daily for 3–5 days) are followed by oral corticosteroid taper. Cyclophosphamide has traditionally been used for severe, rapidly progressive disease. Rituximab (anti-CD20 monoclonal antibody) is increasingly used as an alternative to cyclophosphamide, particularly for renal-limited disease and in patients where cyclophosphamide toxicity is a concern. Plasmapheresis should be considered for patients with severe pulmonary hemorrhage or rapidly progressive renal failure with creatinine >5.8 mg/dL or oliguria.
Anti-GBM Disease
This disease requires the most urgent treatment of all forms of acute GN. High-dose corticosteroids (IV methylprednisolone) should be initiated immediately. Plasmapheresis is essential to remove circulating anti-GBM antibodies; daily plasmapheresis should continue until anti-GBM antibodies become undetectable. Cyclophosphamide is added to suppress ongoing antibody production. The combination of corticosteroids, plasmapheresis, and cyclophosphamide can halt disease progression if initiated early, but delayed treatment results in permanent renal failure.
Emergency Alert — Anti-GBM Disease: If the patient is already dialysis-dependent at diagnosis, renal recovery is extremely rare. Treatment must be initiated within hours, not days. Every hour of delay reduces the chance of renal recovery.
Lupus Nephritis
Corticosteroids form the foundation of therapy. IV methylprednisolone pulses are followed by oral corticosteroid taper. Cyclophosphamide has traditionally been used for severe proliferative disease. Mycophenolate mofetil is increasingly used as an alternative and for maintenance therapy. Azathioprine may be used for long-term maintenance. Hydroxychloroquine should be continued in all patients with SLE for its disease-modifying and renoprotective effects.
Renal Replacement Therapy
Dialysis is indicated when medical management fails to control life-threatening complications of acute kidney injury. Specific indications include: severe azotemia (BUN >100 mg/dL), refractory hyperkalemia (K⁺ >6.5 mEq/L), severe volume overload with pulmonary edema unresponsive to diuretics, symptomatic uremia (encephalopathy, pericarditis, bleeding), and severe metabolic acidosis (pH <7.1). An important clinical distinction is that dialysis in acute GN is often temporary — as inflammation resolves and glomerular function recovers, dialysis can often be discontinued.Section 8
Prognosis and Long-Term Outcomes
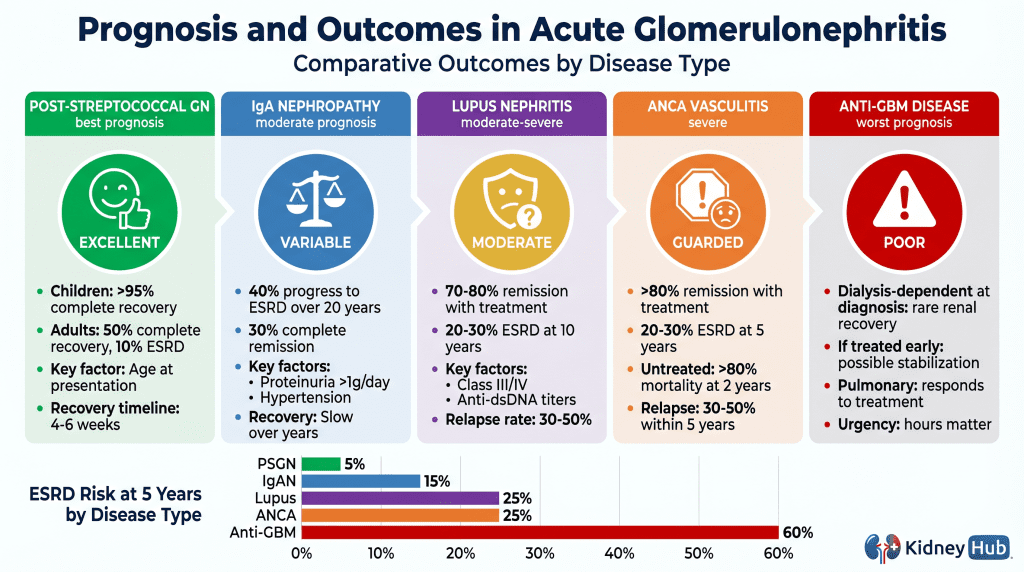
{kind=link}
Post-Streptococcal GN
The prognosis varies significantly by age. In children, more than 95% achieve complete recovery with supportive care alone. In adults, outcomes are less favorable: approximately 50% achieve complete recovery, 20% have partial recovery with persistent mild proteinuria or reduced GFR, 20% develop chronic glomerulonephritis, and 10% progress to ESRD. Predictors of poor prognosis include older age, severe renal impairment at presentation, and extensive crescent formation on biopsy.
IgA Nephropathy
The course is variable and often indolent. Approximately 40% of patients progress to ESRD over 20 years. Predictors of poor prognosis include proteinuria exceeding 1 g/day, hypertension, reduced GFR at presentation, and advanced histological grade (Oxford MEST-C score). ACE inhibitors and ARBs slow progression. Immunosuppressive therapy benefits selected patients with severe disease.
ANCA-Associated Vasculitis
Without treatment, more than 80% of patients die within 2 years. With modern immunosuppressive therapy, remission rates exceed 80%. However, relapse is common, occurring in 30–50% of patients within 5 years. Regarding renal outcomes, 10–20% of patients are dialysis-dependent at presentation; over 5 years, 20–30% develop ESRD. Factors predicting poor renal outcomes include older age, severe renal impairment at presentation, and extensive crescent formation.
Anti-GBM Disease
This disease carries the worst renal prognosis among all forms of acute GN. If the patient is dialysis-dependent at the time of diagnosis, renal recovery is rare. If treated before dialysis dependence develops, renal function may stabilize or improve. The prognosis for pulmonary hemorrhage is better — most patients survive the acute episode with appropriate treatment, though pulmonary function may be permanently impaired. Recurrence is rare but has been reported after renal transplantation if anti-GBM antibodies persist.
Lupus Nephritis
Prognosis depends on the histological class and severity of disease. With modern immunosuppressive therapy, most patients achieve remission or significant improvement. However, renal relapses occur in 30–50% of patients. Long-term renal survival depends on achieving and maintaining remission of proteinuria. Patients with lupus nephritis also carry significantly elevated cardiovascular risk from chronic inflammation, hypertension, and immunosuppressive therapy.Section 9
Complications of Acute Glomerulonephritis
Acute Phase Complications
Hypertensive Emergencies
Severe hypertension can cause hypertensive encephalopathy with headache, altered mental status, seizures, and loss of consciousness. Acute coronary syndrome can develop due to increased myocardial oxygen demand. Acute heart failure with pulmonary edema can develop from volume overload and hypertension. Prompt blood pressure control is essential to prevent end-organ damage.
Pulmonary Complications
Pulmonary edema manifests as dyspnea, orthopnea, and paroxysmal nocturnal dyspnea. In severe cases, acute respiratory distress syndrome (ARDS) develops. In pulmonary-renal syndromes (Goodpasture syndrome, ANCA vasculitis), pulmonary hemorrhage causes hemoptysis, severe hypoxemia, and respiratory failure requiring mechanical ventilation — a life-threatening emergency.
Metabolic Complications
Severe hyperkalemia (K⁺ >6.5 mEq/L) causes cardiac arrhythmias and can lead to cardiac arrest. Severe metabolic acidosis (pH <7.1) impairs cardiac function and cellular metabolism. Hypocalcemia can cause tetany and seizures. These electrolyte emergencies may require urgent dialysis.
Rapidly Progressive Renal Failure
Fulminant acute kidney injury with rapid rise in creatinine indicates extensive glomerular injury. Crescent formation in more than 50% of glomeruli predicts poor renal outcome without urgent immunosuppressive therapy. Oliguria or anuria indicates severe renal dysfunction requiring urgent dialysis.
Long-Term Complications
Chronic Kidney Disease and ESRD
Persistent proteinuria and hypertension after the acute phase indicate ongoing glomerular injury. Progressive decline in GFR over months to years reflects glomerulosclerosis and tubular atrophy. Approximately 10–30% of patients with acute GN progress to ESRD requiring long-term dialysis or transplantation, with the percentage varying significantly by etiology. Disease recurrence in transplanted kidneys occurs in 20–40% of patients with IgA nephropathy and ANCA-associated vasculitis.
Cardiovascular Disease
Patients with acute GN have accelerated atherosclerosis and increased cardiovascular mortality. Left ventricular hypertrophy develops from chronic hypertension. Myocardial infarction and stroke risk are significantly elevated compared to the general population. Cardiovascular risk management — including blood pressure control, lipid management, and smoking cessation — is an essential component of long-term care.Section 10
Clinical Pearls and Key Points
- 1Recognition of the acute nephritic syndrome — hematuria with RBC casts, proteinuria, hypertension, edema, and azotemia — should immediately raise suspicion for acute glomerulonephritis and prompt urgent diagnostic evaluation.
- 2Complement levels guide the differential diagnosis: Low C3 with normal C4 suggests PSGN or C3 glomerulopathy; low C3 and C4 suggests lupus nephritis; normal C3 and C4 suggests IgAN, ANCA vasculitis, or anti-GBM disease.
- 3Timing of hematuria relative to infection is diagnostically critical: Hematuria 1–3 weeks after streptococcal infection suggests PSGN; hematuria concurrent with upper respiratory infection suggests IgA nephropathy (synpharyngitic hematuria).
- 4Pulmonary hemorrhage (hemoptysis) in the setting of acute GN indicates pulmonary-renal syndrome — either Goodpasture syndrome or ANCA-associated vasculitis — and represents a medical emergency requiring immediate workup and treatment.
- 5Early kidney biopsy should be considered in adults with suspected acute GN, as the diagnosis is often not “classic” PSGN, and missing diagnoses like ANCA vasculitis or anti-GBM disease can be fatal without prompt immunosuppressive therapy.
- 6Anti-GBM disease is a true medical emergency: If the patient is already dialysis-dependent at diagnosis, renal recovery is rare. Plasmapheresis must be initiated within hours. Every hour of delay reduces the chance of renal recovery.
- 7Blood pressure control is the most effective long-term intervention to slow progression to chronic kidney disease, regardless of the underlying etiology. ACE inhibitors and ARBs are preferred for their additional renoprotective effects.
- 8Dialysis in acute GN is often temporary: as inflammation resolves and glomerular function recovers, dialysis can frequently be discontinued — a critical distinction from chronic kidney disease requiring permanent renal replacement therapy.
- 9Persistent proteinuria after the acute phase is the most important predictor of progression to chronic kidney disease and should be monitored closely and treated aggressively with ACE inhibitors or ARBs.
- 10Long-term follow-up is essential even after apparent clinical recovery, as some patients develop chronic kidney disease years later. Annual monitoring of blood pressure, urinalysis, and serum creatinine is recommended for all patients with a history of acute GN.
Section 11
Summary
Acute glomerulonephritis represents a diverse group of rapidly progressive kidney diseases with varied etiologies and immunological mechanisms. The acute nephritic syndrome — characterized by hematuria with red blood cell casts, proteinuria, hypertension, edema, and azotemia — is the hallmark clinical presentation that should prompt immediate diagnostic evaluation.
The major etiologies can be classified by immunopathological mechanism: immune complex-mediated forms (PSGN, IgA nephropathy, lupus nephritis, MPGN), pauci-immune ANCA-associated vasculitis (GPA, MPA, EGPA), and anti-GBM disease (with or without pulmonary involvement as Goodpasture syndrome). Each has distinctive clinical features, complement patterns, serological markers, biopsy findings, and treatment requirements.
Diagnosis relies on clinical presentation, systematic serological testing guided by complement levels, and kidney biopsy for definitive histological characterization. Management involves universal supportive care — fluid and sodium restriction, diuretics, blood pressure control — combined with etiology-specific therapy ranging from antibiotics alone (PSGN in children) to urgent plasmapheresis and cyclophosphamide (anti-GBM disease).
Early recognition and treatment are critical determinants of renal outcomes. Prognosis varies significantly by etiology, from excellent in children with PSGN (>95% complete recovery) to poor in anti-GBM disease (~60% ESRD at 5 years). Long-term follow-up is essential, as some patients develop chronic kidney disease years after apparent clinical recovery. The field continues to evolve with newer targeted therapies — particularly rituximab for ANCA vasculitis and emerging complement inhibitors for C3 glomerulopathy — offering improved outcomes with reduced toxicity.
References
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney International. 2021;100(4S):S1–S276.
- Couser WG. Rapidly progressive glomerulonephritis: Classification, pathophysiology, diagnosis, and management. American Journal of Kidney Diseases. 2021;78(3):389–407.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis & Rheumatism. 2013;65(1):1–11.
- Floege J, Feehally J, Tonelli M (Eds.). Comprehensive Clinical Nephrology, 6th ed. Philadelphia: Elsevier; 2019.
- Sethi S, Fervenza FC. Pathology of renal diseases associated with dysfunction of the alternative complement pathway. Kidney International. 2022;102(1):5–17.
- Fogo AB. AJKD Atlas of Renal Pathology: Postinfectious Glomerulonephritis. American Journal of Kidney Diseases. 2015;67(2):e17–e18.
- Chadban SJ, Atkins RC. Glomerulonephritis. The Lancet. 2005;365(9473):1797–1806.
- Nasr SH, Markowitz GS. Acute Postinfectious Glomerulonephritis. In: Comprehensive Clinical Nephrology, 5th ed. Philadelphia: Elsevier; 2016:312–325.
- Appel GB, Radhakrishnan J, D’Agati VD. Secondary Glomerulonephritis. In: Brenner and Rector’s The Kidney, 10th ed. Philadelphia: Elsevier; 2016:1268–1349.
- Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nature Reviews Nephrology. 2012;8(11):634–642.
Kidney Hub · Chapter 6: Acute Glomerulonephritis · Educational Material
All content is for educational purposes only. Clinical decisions should be based on individual patient assessment and current guidelines.
© 2026 Kidney Hub · Evidence-Based Nephrology Education